|
|

|
|
|
Computing Speed 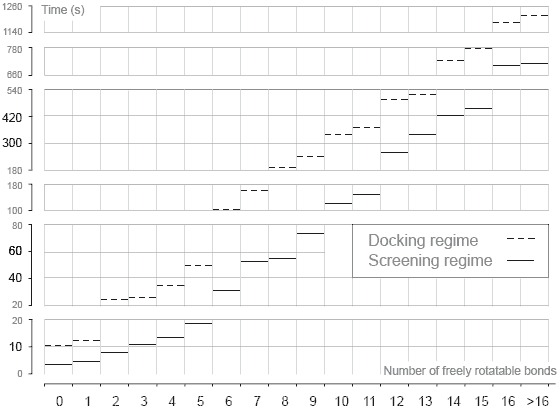 The speed of ligand docking calculations depends on the settings of genetic algorithm search. Currently, Lead Finder provides two types of calculation regime: a more accurate and exhaustive search in the default docking regime and a faster search at the cost of slight decrease of accuracy in the screening regime. While the setting for the default docking settings have been adjusted to achieve maximum docking accuracy, the settings for the screening regime were fine-tuned to achieve maximum speed of calculations at the cost of slight (~5%) decrease in Docking Success Rate.
To benchmark the speed of docking calculations, a set of 407 diverse protein-ligand complexes (used also for Docking Success Rate measurements) was used. All calculations were performed in two regimes: docking and screening. The above figure presents results of the current benchmarking study with average times needed to dock a ligand as a function of ligand's degrees of freedom (number of freely rotatable bonds). The screening regime is 2-4 times faster than the docking regime. In screening regime, Lead Finder docks a ligand with 6 freely rotatable bonds within 30 sec on average. |
|
a) Time pre ligand indicates average time (in seconds) of docking one compound from the library (on the single processor core).
b) Total screening time total time (in hours) of virtual library screening on the computer cluster. c) Volume of the energy grid indicates the volume of the energy grid (for more details on grid definition see Docking algorithm section). We also benchmarked the speed of ligand docking in a number of real-life
virtual screening studies. In one study, a commercial library of 300,000 compounds
(STK library by Vitas-M Laboratory)
was screened against a number of protein targets. Using a cluster of 16 computational
nodes (dual Xeon 5150 2.6 GHz) we were able to screen the entire
library in 12 to 24 hours depending on the protein target. The average time for docking
of a compound from the library for a particular protein is provided in the table above.
To estimate relative speed of docking calculations in different computing environments, the following table can be used as a guide:
|
|
|
|
|
E-mail: info@biomoltech.com |
Phone: +1(416)238-1263 |
Fax: +1(416)352-6117 Mailing address: 226 York Mills Rd, Toronto, Ontario M2L 1L1, Canada |