|
Lead Finder's Main Application Areas
Docking of small molecules to protein structures and evaluation of parameters of such
molecular interaction may provide useful information to researchers in many life science areas. However,
virtual screening, ligand docking and estimation of binding energy of protein-ligand
interaction are clearly the three primary application areas for Lead Finder. Additionally,
Lead Finder can be used to prepare protein structures for independent docking experiments.
- Virtual Screening
Lead Finder screens large libraries of chemical compounds against a protein target
to find potent binders with high fidelity with a typical speed of
5,000 compounds per processor/core per day. The ability of Lead Finder to find active
compounds in mixtures with inactive ones has been extensively validated on a set
of 34 therapeutically relevant protein targets, showing impressive enrichment results
in most cases.
- Ligand Docking
Lead Finder correctly predicts 3D structure of non-covalently and covalently bound
protein-ligand complexes in most cases. Accuracy of protein-ligand docking has been validated on a set
of 407 protein-ligand complexes, which appears to be the most extensive
benchmarking study of such kind at present. Our test set was a combination of test sets of such
molecular docking software as
FlexX,
Glide SP, Glide XP,
Gold,
LigandFit,
MolDock,
Surflex.
With this test set, we attempted a straightforward comparison of Lead Finder's performance to that of
competitive programs. As shown in our benchmarking study, Lead Finder has
outperformed all competitive programs on their native test sets.
- Binding Energy Estimation
Lead Finder performs high-accuracy estimations of the free energy of protein-ligand
binding based on an original semi-empiric molecular-mechanical scoring function.
The accuracy of binding energy estimations has been validated on a set of experimentally
measured binding energies for 330 diverse protein-ligand complexes, which appears to be
the most extensive benchmarking study of such kind at present. The root mean square deviation (RMSD) of Lead Finder's
predicted values from the experimental values of binding energies has been less than 1.50 kcal/mol in most cases,
which appears to be the highest accuracy among the competitive docking programs at present.
- Protein Structure Preparation
Lead Finder automatically prepares fully functional protein structures
(for molecular docking experiments and other modeling purposes) starting from crude heavy
atom coordinates (as usually present in PDB files and homology models) by adding
hydrogen atoms to protein residues (ligands, substrates, cofactors) at a given
pH. An original electrostatic model has been implemented in Lead Finder for accurate
prediction of ionization states for aminoacid residues and validated on set of
100 diverse pKa values of ionizable residues.
 |
|
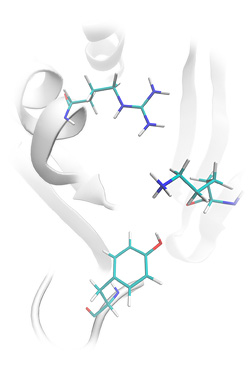
Tyrosine phosphatase structure prepared for ligand docking: Lead Finder restored missing amino acids side chains (not resolved in the original pdb structure), calculated the ionization state of the protein and added hydrogen atoms accordingly.
|
|
|