|
Docking Algorithm
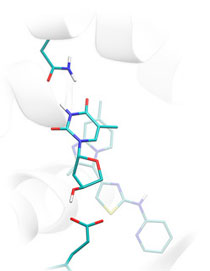
From the mathematical point of view, ligand docking involves search of the global minimum on the multidimensional surface describing the free energy of protein-ligand binding. With ligands possessing 20-30 degrees of freedom, including translational and rotational degrees of freedom and the freely rotatable bonds, and complex nature of the energy surface that involves multiple energy terms, the deterministic global optimum search is presently not possible by practical means. To tackle this computationally challenging problem, Lead Finder uses its own approach of combining the genetic algorithm search with local optimization procedures and smart exploitation of the knowledge generated during a search run. Rational combination of different optimization strategies makes Lead Finder efficient in coarse sampling of ligands phase space and following refinement of promising solutions.
Lead Finder's docking algorithm can be fine-tuned by end user through configuration settings. There are a few dozen configuration settings available, each of which makes a certain impact on the algorithm performance, robustness and speed of calculations. For simplicity and ease of use, two calculation regimes have been introduced in Lead Finder: docking regime and screening regime. The default docking regime provides maximum docking accuracy at a slower speed of output. The screening regime has been designed to accelerate computing performance while minimizing loss of docking accuracy. More details about the accuracy and speed of docking calculations in docking and screening regimes can be found in the relevant pages in the Science section.
The more detailed description of the Lead Finder docking algorithm is available as a PDF document. |